Stephen Muench
- Position: Associate Professor
- Areas of expertise: Structural Biology; electron microscopy; membrane protein.
- Email: S.P.Muench@leeds.ac.uk
- Phone: +44(0)113 343 4279
- Website: Twitter | Googlescholar
Profile
I studied for an undergraduate degree in Biochemistry and Microbiology at the University of Sheffield in 1997 during which time I undertook an undergraduate research project in X-ray crystallography, which fostered my strong interest in structural biology. Therefore, I continued in Sheffield for my PhD studies looking into the development of new anti-malarial and toxoplasmosis compounds through X-ray crystallography. After a brief postdoc in Sheffield studying the role of the GTPAse EngA I moved to Leeds in 2005 to learn electron microscopy (EM). During this time I was able to develop a keen interest in EM and its use for studying large membrane protein complexes. After the award of an MRC career development fellowship in 2010 I was able to start my own group with a strong interest in combining different techniques to study the structure and function of a wide range of protein targets with the overall aim of driving structure based inhibitor design.
I am currently a Senior lecturer in membrane biology at the University of Leeds. My group has worked on a range of systems from ion channels to developing new biosensors. Recent, major contributions to the field include: (i) The use of EM to drive inhibitor design for the membrane bound bc1 complex, (ii) The first negative stain and cryo-EM reconstruction of a membrane protein extracted using styrene maleic acid lipid particles (SMALPs), which removes the need for detergents. (iii) The first use of tagging to identify the binding site of small molecule inhibitors using EM. (iv) A de novo built EM structure with an inhibitor bound which reveals the binding site and co-ordination with neighboring metal atoms. (v) work on new hybrid vesicles with block co-polymers to improve membrane protein lifetimes. (vi) The first cryo-EM structure of the V-ATPase complex and the first sub nm V-ATPase reconstruction, providing new insights into the structure and mechanism of this important membrane bound proton pump.
Research interests
Overall Goal
One of the major challenges for structural biology is not just to resolve the structure of large protein complexes, but crucially also to appreciate the dynamics and conformational variability of such systems. These challenges are best met through a combination of techniques such as X-ray crystallography and Electron Microscopy (EM), in particular time-resolved applications. My research involves the combination of these techniques in order to understand the structure/function relationship of a number of medically important targets, with a particular interest in membrane proteins and use this information to underpin the design of small molecule inhibitors.
Structure, function and molecular mechanism of the vacuolar ATPase
The vacuolar H+-ATPase (V-ATPase) is an ATP-driven proton pump essential for the function of virtually all eukaryotic cells. It is a membrane-bound rotary motor, larger and more complex than the related ATPase synthase, and is responsible for acidifying intracellular compartments, energising membrane transport and pH homeostasis. Mutations in V-ATPase genes cause osteopetrosis, kidney disease and autophagic myopathy, and its involvement in osteoporosis and metastatic cancer means that controlling its activity with drugs has therapeutic potential.
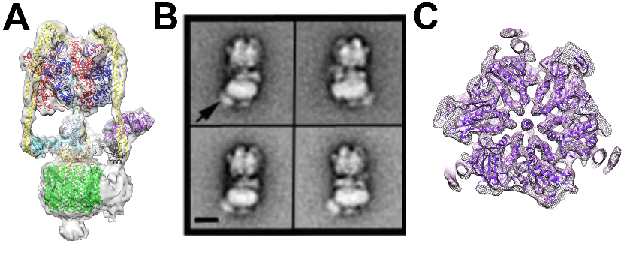
Figure1. A) M. sexta V-ATPase single particle cryo-EM reconstruction with subunit homologues fitted, B) Negative stain analysis of the V-ATPase bound to a Pa1b-Strep-HRP complex, highlighted by an arrow (scale bar is 10nm). C) Slice through of the EM map of the catalytic V1 domain after accounting for flexibility in the complex.
Despite its importance, our understanding of the V-ATPase mechanism and regulation is poor and is a major block in therapeutic development, since structural data often underpins therapeutic drug design. We published the first sub nm structure of the V-ATPase by single particle cryo-EM (Figure 1A), providing new insights into its structure and mechanism. Moreover, using a novel tagging mechanism we have studied the binding of the selective inhibitor PA1b to the V-ATPase (Figure 1B). Through accounting for the inherent flexibility within the system, in particular of the V1 and Vo domains its also possible improve the resulting resolution of the EM reconstruction (Figure 1C). This complex motor is regulated in response to physiological signals indicating low energy, leading to reversible domain dissociation. The nature of the structural changes that occur in response to these signals is being studied by single particle cryo-EM and, where appropriate, crystallography.
Structure Based Drug Design
In addition to working on the V-ATPase we are working on a number of therapeutic targets both membrane bound and soluble and ranging from 120kDa->1MDa. Through single particle EM studies we have now been able to directly visualise the small molecule inhibitor opening up new avenues for structure based drug design on systems, intractable to other structural techniques. We have used an EM approach to understand the difference in inhibitor potency between yeast and Arabidopsis Imidazoleglycerol-phosphate dehydratase (IGPD)(Figure 2A) . Moreover, the group, in collaboration with the University of Liverpool have used EM to study the binding of inhibitors to cytochrome bc1, a validated antimalarial target (Figure 2B).
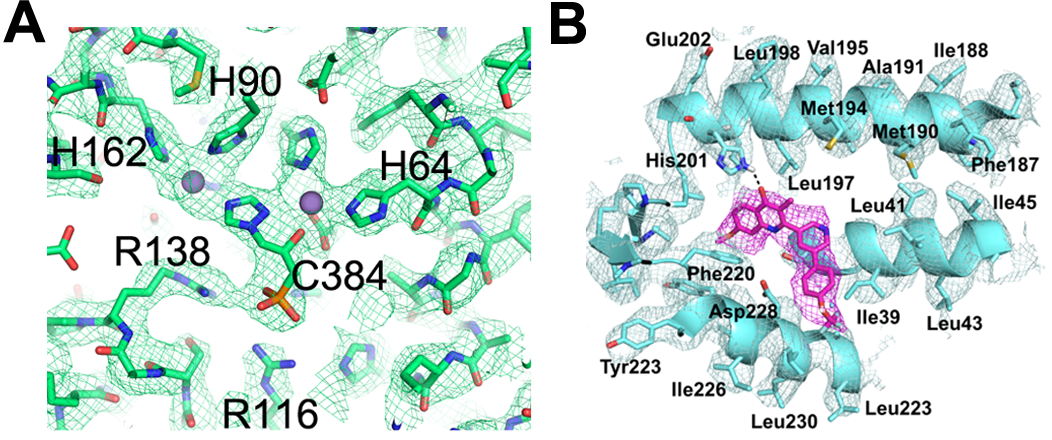
Figure 2 (A) Inhibitor C384 bound within the yeast Imidazoleglycerol-phosphate dehydratase (IGPD) as visualised in the single particle cryo-EM map. (B) Single particle cryo-EM map of bovine cytochrome bc1 complex (blue) with small molecule inhibitor bound (magenta).
Developing new ways to study membrane proteins
Despite the biological importance of membrane proteins and their relevance for drug targeting with over >60% of therapeutic targets being membrane proteins, our structural understanding of this class of proteins is much poorer than that of their soluble counterparts. There are a number of challenges associated with studying membrane proteins including difficulties in protein expression and the requirement to stabilise the protein outside of the membrane environment, usually with detergents. The group have been looking at the ability of styrene maleic acid (SMA) copolymers to extract membrane proteins from their lipid environment surrounded by more native lipid. This has been shown to improve stability of the membrane protein and the group published the first negative stain and sub nm single particle cryo-EM structure of an SMA extracted membrane protein. Future work is combining EM with mass spectrometry to study the role of native lipids in the stability of membrane proteins and the effect of overexpression on the native lipid environment. This work is through both academic and industrial collaborations.
<h4>Research projects</h4> <p>Some research projects I'm currently working on, or have worked on, will be listed below. Our list of all <a href="https://medicinehealth.leeds.ac.uk/dir/research-projects">research projects</a> allows you to view and search the full list of projects in the faculty.</p>Co-investigator (Co-I)
Research groups and institutes
- Multidisciplinary Cardiovascular Research Centre