Amelogenesis Research Group
- Group summary: The group aims to understand tooth, and specifically enamel, formation and function by studying conditions such as Amelogenesis Imperfecta, an inherited failure of enamel development.
Mission statement
The Leeds Amelogenesis Research Group aims to increase our understanding of enamel formation and function. To do this, we aim to identify and study the mechanisms that lead to conditions such as Amelogenesis Imperfecta, Molar Incisor Hypomineralisation Dental Fluorosis and Dental Caries, as well as studying the normal processes involved in enamel development and the evolutionary history of enamel.
What is Amelogenesis Imperfecta (AI)?
Amelogenesis Imperfecta (AI) is a variable group of inherited conditions of tooth enamel, in which both the quality and the quantity of enamel may be abnormal. AI mostly occurs in the absence of other health problems (non-syndromic AI), but can also be associated with significant problems in other tissues or organs (syndromic AI). This can include problems with the skin, kidneys, hearing, vision and immunity to name only a few.
The impact of AI on affected individuals and their families is considerable. Patients experience dental pain and sensitivity, poor aesthetics and are reported to experience a lower quality of life due to social anxiety. Treatment of AI is expensive and difficult since it requires ongoing clinical management to maintain function and aesthetics. AI affects around 1 in 2000 people globally and can be inherited in an autosomal dominant, recessive or X-linked manner.
Amelogenesis Imperfecta
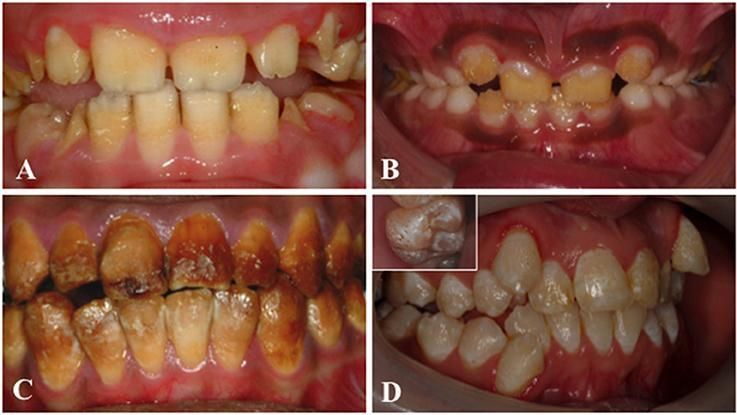
Clinical images that illustrate the variability of AI. (A) Hypoplastic AI is characterised by teeth without the curves associated with a normal enamel volume. (B) In hypomaturation AI enamel volume can be near-normal, but opaque with structural weaknesses that result in rapid post-eruptive enamel loss with enamel fracturing away to exposure the underlying dentine. (C) Brown discolouration and early post-eruptive enamel loss is typical of hypomineralised forms of AI. (D) Mixed AI phenotypes are frequently encountered. In this example a near-normal enamel volume is characterised by multiple focal pits that are most evident on the inset image, with variable colouration that includes focal opacities, but without premature fracturing of the enamel to reveal dentine. Image reused with acknowledgement to Front. Physiol., 26 June 2017 | https://doi.org/10.3389/fphys.2017.00435
Whilst AI is classed as a “rare” disease, studying AI has the potential to unlock information about how enamel forms and fails, which is relevant to the study, and potentially also the treatment, of other more common enamelopathies (clinically relevant forms of enamel failure) which together impact >2.5 billion people around the world, are an immense societal and healthcare burden and disproportionately impact socially disadvantaged groups.
Our Research
The Amelogenesis Research Group uses various sequencing methodologies, including exome and genome sequencing and downstream analyses to identify the underlying AI mutations in patients with amelogenesis imperfecta. We collect both DNA samples and teeth from patients and their families, via an ever-expanding Clinical Excellence Network of over 60 paediatric dentists across all nations of the UK. We also collaborate with international clinicians and researchers including in France, Costa Rica, Venezuela, Oman and Sudan. We have DNA from over 500 AI families, making our AI research cohort one of the largest in the world.
We utilise well-equipped state-of-the-art sequencing facilities run by our collaborator Dr Chris Watson and the high capacity, high speed Aire system, provided by Leeds Research Computing, for sequencing analysis.
Through our successful sequencing studies, our group have already implicated a number of genes in AI pathogenesis, including PLXNB2, SP6, GPR68, AMTN, PEX1, PEX6, AMBN, ITGB6, LAMB3, FAM20A, WDR72, C4orf26, SLC24A4 and CNNM4. This knowledge is the first step in elucidating roles for these proteins in enamel formation.
As well as DNA sequencing studies, we were also the first to discover that some types of amelogenesis imperfecta are examples of proteopathies. To further this research and to potentially identify other pathomechanisms for AI, we are creating new cellular models of amelogenesis and amelogenesis imperfecta using human stem cells. With support from the Leeds Centre for Disease Models we have developed techniques to derive ameloblast-like cells from human stem cells. We have also used CRISPR-Cas9 gene editing to specifically engineer variants that cause AI into these cells. We then study the effects of the mutations and are using these models to develop therapeutics that could potentially prevent AI from developing in permanent teeth. Such therapeutics might also be effective in treating linked syndromic disease phenotypes.
We also work to characterise the phenotypes of AI enamel resulting from particular genotypes through collaboration with Prof Maisoon Al-Jawad and the Leeds Biomineralisation Research Group. We use scanning electron microscopy, micro-computerised tomography, energy dispersive X-ray analysis and microhardness testing to characterise the enamel. We also have access to synchrotron X-ray fluorescence analysis at the Diamond Light Source, Harwell. This research will guide clinical management of AI and will help to focus research efforts on improving restorative techniques for the treatment of enamel pathology. We are also developing new methods to study how ameloblasts move during enamel formation to better understand how enamel is made, what goes wrong in AI, to highlight therapeutic targets and to aid development of new restorative treatments.
Research Impact and Public Engagement
Our research has helped to establish UK NHS diagnostic screening for AI, which was used as the basis for the Genomes England R340 Clinical Amelogenesis Imperfecta panel, which we continue to curate to efficiently translate our research findings into clinical impact.
Our group were also instrumental in establishing a UK-wide Clinical Excellence Network for AI and we have also published a comprehensive and highly cited review of AI genetics.
We educate the dentists of the future, Leeds University dental students, about amelogenesis imperfecta at both undergraduate and master’s degree level.
As part of the SMILE AIDER (Stakeholder Meaningful InvoLvement and Engagement Aiding Dental Research) oral health patients and public involvement forum and as international friends of the D3 (Developmental Dental Defects) group, we participate in increasing the public understanding of developmental dental defects such as amelogenesis imperfecta, molar incisor hypomineralisation (MIH) and fluorosis.
As part of our efforts to encourage involvement in dentistry and research, we host undergraduate students for summer project placements and give further education students experience of research in a laboratory setting. We have also participated in Leeds Dental School Open Days and have run an innovative and exciting “Dental Disease Detectives” family activity programme at the Thackray Medical Museum.
Some members of our group are also Science, Technology, Engineering and Mathematics (STEM) ambassadors who volunteer their time, enthusiasm and experiences to encourage and inspire young people to achieve more and progress further in science, technology, engineering and mathematics.
Key publications
1: Smith CEL, Laugel-Haushalter V, Hany U, Best S, Taylor RL, Poulter JA,
Wortmann SB, Feichtinger RG, Mayr JA, Al Bahlani S, Nikolopoulos G, Rigby A,
Black GC, Watson CM, Mansour S, Inglehearn CF, Mighell AJ, Bloch-Zupan A; UK
Inherited Retinal Disease Consortium, Genomics England Research Consortium.
Biallelic variants in Plexin B2 (PLXNB2) cause amelogenesis imperfecta,
hearing loss and intellectual disability. J Med Genet. 2024 Jun
20;61(7):689-698. doi: 10.1136/jmg-2023-109728.
2: Monteiro J, Balmer R, Lafferty F, Lyne A, Mighell A, O'Donnell K, Parekh S.
Establishment of a clinical network for children with amelogenesis imperfecta
and dentinogenesis imperfecta in the UK: 4-year experience. Eur Arch Paediatr
Dent. 2024 Feb;25(1):85-91. doi: 10.1007/s40368-023-00859-2. Epub 2024 Feb 3.
3: Hany U, Watson CM, Liu L, Nikolopoulos G, Smith CEL, Poulter JA, Brown CJ,
Patel A, Rodd HD, Balmer R, Harfoush A, Al-Jawad M, Inglehearn CF, Mighell AJ.
Novel Ameloblastin Variants, Contrasting Amelogenesis Imperfecta Phenotypes. J
Dent Res. 2024 Jan;103(1):22-30. doi: 10.1177/00220345231203694.
4: Hany U, Watson CM, Liu L, Smith CEL, Harfoush A, Poulter JA, Nikolopoulos G,
Balmer R, Brown CJ, Patel A, Simmonds J, Charlton R, Acosta de Camargo MG, Rodd
HD, Jafri H, Antanaviciute A, Moffat M, Al-Jawad M, Inglehearn CF, Mighell AJ.
Heterozygous COL17A1 variants are a frequent cause of amelogenesis
imperfecta. J Med Genet. 2024 Mar 21;61(4):347-355. doi:
10.1136/jmg-2023-109510.
5: Lafferty F, Al Siyabi H, Sinadinos A, Kenny K, Mighell AJ, Monteiro J,
Soldani F, Parekh S, Balmer RC. The burden of dental care in Amelogenesis
Imperfecta paediatric patients in the UK NHS: a retrospective, multi-centred
analysis. Eur Arch Paediatr Dent. 2021 Oct;22(5):929-936. doi:
10.1007/s40368-021-00638-x.
6: Nikolopoulos G, Smith CEL, Poulter JA, Murillo G, Silva S, Lamb T, Berry IR,
Brown CJ, Day PF, Soldani F, Al-Bahlani S, Harris SA, O'Connell MJ, Inglehearn
CF, Mighell AJ. Spectrum of pathogenic variants and founder effects in
amelogenesis imperfecta associated with MMP20. Hum Mutat. 2021
May;42(5):567-576. doi: 10.1002/humu.24187.
7: Sato K, Mogi C, Mighell AJ, Okajima F. A missense mutation of Leu74Pro of
OGR1 found in familial amelogenesis imperfecta actually causes the loss of the
pH-sensing mechanism. Biochem Biophys Res Commun. 2020 Jun 11;526(4):920-926.
doi: 10.1016/j.bbrc.2020.04.005. Epub 2020 Apr 10.
8: Smith CEL, Whitehouse LLE, Poulter JA, Wilkinson Hewitt L, Nadat F, Jackson
BR, Manfield IW, Edwards TA, Rodd HD, Inglehearn CF, Mighell AJ. A missense
variant in specificity protein 6 (SP6) is associated with amelogenesis
imperfecta. Hum Mol Genet. 2020 Jun 3;29(9):1417-1425. doi: 10.1093/hmg/ddaa041.
9: Nikolopoulos G, Smith CEL, Brookes SJ, El-Asrag ME, Brown CJ, Patel A,
Murillo G, O'Connell MJ, Inglehearn CF, Mighell AJ. New missense variants in
RELT causing hypomineralised amelogenesis imperfecta. Clin Genet. 2020
May;97(5):688-695. doi: 10.1111/cge.13721.
10: Smith CEL, Poulter JA, Brookes SJ, Murillo G, Silva S, Brown CJ, Patel A,
Hussain H, Kirkham J, Inglehearn CF, Mighell AJ. Phenotype and Variant Spectrum
in the LAMB3 Form of Amelogenesis Imperfecta. J Dent Res. 2019
Jun;98(6):698-704. doi: 10.1177/0022034519835205.
11: McDowall F, Kenny K, Mighell AJ, Balmer RC. Genetic testing for amelogenesis
imperfecta: knowledge and attitudes of paediatric dentists. Br Dent J. 2018 Aug
24;225(4):335-339. doi: 10.1038/sj.bdj.2018.641.
12: Whitehouse LLE, Smith CEL, Poulter JA, Brown CJ, Patel A, Lamb T, Brown LR,
O'Sullivan EA, Mitchell RE, Berry IR, Charlton R, Inglehearn CF, Mighell AJ.
Novel DLX3 variants in amelogenesis imperfecta with attenuated tricho-dento-
osseous syndrome. Oral Dis. 2019 Jan;25(1):182-191. doi: 10.1111/odi.12955. Epub
2018 Sep 9.
13: Smith CEL, Poulter JA, Antanaviciute A, Kirkham J, Brookes SJ, Inglehearn
CF, Mighell AJ. Amelogenesis Imperfecta; Genes, Proteins, and Pathways. Front
Physiol. 2017 Jun 26;8:435. doi: 10.3389/fphys.2017.00435.
14: Smith CEL, Kirkham J, Day PF, Soldani F, McDerra EJ, Poulter JA, Inglehearn
CF, Mighell AJ, Brookes SJ. A Fourth <i>KLK4</i> Mutation Is Associated with
Enamel Hypomineralisation and Structural Abnormalities. Front Physiol. 2017 May
29;8:333. doi: 10.3389/fphys.2017.00333.
15: Smith CE, Whitehouse LL, Poulter JA, Brookes SJ, Day PF, Soldani F, Kirkham
J, Inglehearn CF, Mighell AJ. Defects in the acid phosphatase ACPT cause
recessive hypoplastic amelogenesis imperfecta. Eur J Hum Genet. 2017
Aug;25(8):1015-1019. doi: 10.1038/ejhg.2017.79.
16: Brookes SJ, Barron MJ, Smith CEL, Poulter JA, Mighell AJ, Inglehearn CF,
Brown CJ, Rodd H, Kirkham J, Dixon MJ. Amelogenesis imperfecta caused by
N-terminal enamelin point mutations in mice and men is driven by endoplasmic
reticulum stress. Hum Mol Genet. 2017 May 15;26(10):1863-1876. doi:
10.1093/hmg/ddx090.
17: Parry DA, Smith CE, El-Sayed W, Poulter JA, Shore RC, Logan CV, Mogi C, Sato
K, Okajima F, Harada A, Zhang H, Koruyucu M, Seymen F, Hu JC, Simmer JP, Ahmed
M, Jafri H, Johnson CA, Inglehearn CF, Mighell AJ. Mutations in the pH-Sensing
G-protein-Coupled Receptor GPR68 Cause Amelogenesis Imperfecta. Am J Hum Genet. 2016 Oct 6;99(4):984-990. doi: 10.1016/j.ajhg.2016.08.020.
18: Smith CE, Murillo G, Brookes SJ, Poulter JA, Silva S, Kirkham J, Inglehearn
CF, Mighell AJ. Deletion of amelotin exons 3-6 is associated with amelogenesis
imperfecta. Hum Mol Genet. 2016 Aug 15;25(16):3578-3587. doi:
10.1093/hmg/ddw203.
19: Smith CE, Poulter JA, Levin AV, Capasso JE, Price S, Ben-Yosef T, Sharony R,
Newman WG, Shore RC, Brookes SJ, Mighell AJ, Inglehearn CF. Spectrum of PEX1 and
PEX6 variants in Heimler syndrome. Eur J Hum Genet. 2016 Nov;24(11):1565-1571.
doi: 10.1038/ejhg.2016.62.
20: Poulter JA, Smith CE, Murrillo G, Silva S, Feather S, Howell M, Crinnion L,
Bonthron DT, Carr IM, Watson CM, Inglehearn CF, Mighell AJ. A distinctive oral
phenotype points to FAM20A mutations not identified by Sanger sequencing. Mol
Genet Genomic Med. 2015 Oct 4;3(6):543-9. doi: 10.1002/mgg3.164.
21: Ratbi I, Falkenberg KD, Sommen M, Al-Sheqaih N, Guaoua S, Vandeweyer G,
Urquhart JE, Chandler KE, Williams SG, Roberts NA, El Alloussi M, Black GC,
Ferdinandusse S, Ramdi H, Heimler A, Fryer A, Lynch SA, Cooper N, Ong KR, Smith
CE, Inglehearn CF, Mighell AJ, Elcock C, Poulter JA, Tischkowitz M, Davies SJ,
Sefiani A, Mironov AA, Newman WG, Waterham HR, Van Camp G. Heimler Syndrome Is
Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and
PEX6. Am J Hum Genet. 2015 Oct 1;97(4):535-45. doi: 10.1016/j.ajhg.2015.08.011.
22: Acevedo AC, Poulter JA, Alves PG, de Lima CL, Castro LC, Yamaguti PM, Paula
LM, Parry DA, Logan CV, Smith CE, Johnson CA, Inglehearn CF, Mighell AJ.
Variability of systemic and oro-dental phenotype in two families with non-lethal
Raine syndrome with FAM20C mutations. BMC Med Genet. 2015 Feb 21;16:8. doi:
10.1186/s12881-015-0154-5.
23: de la Dure-Molla M, Quentric M, Yamaguti PM, Acevedo AC, Mighell AJ, Vikkula
M, Huckert M, Berdal A, Bloch-Zupan A. Pathognomonic oral profile of Enamel
Renal Syndrome (ERS) caused by recessive FAM20A mutations. Orphanet J Rare Dis.
2014 Jun 14;9:84. doi: 10.1186/1750-1172-9-84.
24: Poulter JA, Murillo G, Brookes SJ, Smith CE, Parry DA, Silva S, Kirkham J,
Inglehearn CF, Mighell AJ. Deletion of ameloblastin exon 6 is associated with
amelogenesis imperfecta. Hum Mol Genet. 2014 Oct 15;23(20):5317-24. doi:
10.1093/hmg/ddu247.
25: Poulter JA, Brookes SJ, Shore RC, Smith CE, Abi Farraj L, Kirkham J,
Inglehearn CF, Mighell AJ. A missense mutation in ITGB6 causes pitted
hypomineralized amelogenesis imperfecta. Hum Mol Genet. 2014 Apr
15;23(8):2189-97. doi: 10.1093/hmg/ddt616. Epub 2013 Dec 6.
26: Poulter JA, El-Sayed W, Shore RC, Kirkham J, Inglehearn CF, Mighell AJ.
Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as
a cause of dominant hypoplastic amelogenesis imperfecta. Eur J Hum Genet. 2014
Jan;22(1):132-5. doi: 10.1038/ejhg.2013.76. Epub 2013 May 1.
27: Jaureguiberry G, De la Dure-Molla M, Parry D, Quentric M, Himmerkus N, Koike
T, Poulter J, Klootwijk E, Robinette SL, Howie AJ, Patel V, Figueres ML,
Stanescu HC, Issler N, Nicholson JK, Bockenhauer D, Laing C, Walsh SB, McCredie
DA, Povey S, Asselin A, Picard A, Coulomb A, Medlar AJ, Bailleul-Forestier I,
Verloes A, Le Caignec C, Roussey G, Guiol J, Isidor B, Logan C, Shore R, Johnson
C, Inglehearn C, Al-Bahlani S, Schmittbuhl M, Clauss F, Huckert M, Laugel V,
Ginglinger E, Pajarola S, Spartà G, Bartholdi D, Rauch A, Addor MC, Yamaguti PM,
Safatle HP, Acevedo AC, Martelli-Júnior H, dos Santos Netos PE, Coletta RD,
Gruessel S, Sandmann C, Ruehmann D, Langman CB, Scheinman SJ, Ozdemir-Ozenen D, Hart TC, Hart PS, Neugebauer U, Schlatter E, Houillier P, Gahl WA, Vikkula M,
Bloch-Zupan A, Bleich M, Kitagawa H, Unwin RJ, Mighell A, Berdal A, Kleta R.
Nephrocalcinosis (enamel renal syndrome) caused by autosomal recessive FAM20A
mutations. Nephron Physiol. 2012;122(1-2):1-6. doi: 10.1159/000349989.
28: Parry DA, Poulter JA, Logan CV, Brookes SJ, Jafri H, Ferguson CH, Anwari BM,
Rashid Y, Zhao H, Johnson CA, Inglehearn CF, Mighell AJ. Identification of
mutations in SLC24A4, encoding a potassium-dependent sodium/calcium exchanger,
as a cause of amelogenesis imperfecta. Am J Hum Genet. 2013 Feb 7;92(2):307-12.
doi: 10.1016/j.ajhg.2013.01.003.
29: Parry DA, Brookes SJ, Logan CV, Poulter JA, El-Sayed W, Al-Bahlani S, Al
Harasi S, Sayed J, Raïf el M, Shore RC, Dashash M, Barron M, Morgan JE, Carr IM,
Taylor GR, Johnson CA, Aldred MJ, Dixon MJ, Wright JT, Kirkham J, Inglehearn CF,
Mighell AJ. Mutations in C4orf26, encoding a peptide with in vitro
hydroxyapatite crystal nucleation and growth activity, cause amelogenesis
imperfecta. Am J Hum Genet. 2012 Sep 7;91(3):565-71. doi:
10.1016/j.ajhg.2012.07.020.
30: El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. Hypomaturation
amelogenesis imperfecta due to WDR72 mutations: a novel mutation and
ultrastructural analyses of deciduous teeth. Cells Tissues Organs.
2011;194(1):60-6. doi: 10.1159/000322036.
31: El-Sayed W, Shore RC, Parry DA, Inglehearn CF, Mighell AJ. Ultrastructural
analyses of deciduous teeth affected by hypocalcified amelogenesis imperfecta
from a family with a novel Y458X FAM83H nonsense mutation. Cells Tissues Organs.
2010;191(3):235-9. doi: 10.1159/000252801.
32: El-Sayed W, Parry DA, Shore RC, Ahmed M, Jafri H, Rashid Y, Al-Bahlani S, Al
Harasi S, Kirkham J, Inglehearn CF, Mighell AJ. Mutations in the beta propeller
WDR72 cause autosomal-recessive hypomaturation amelogenesis imperfecta. Am J Hum Genet. 2009 Nov;85(5):699-705. doi: 10.1016/j.ajhg.2009.09.014.
33: Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, Dollfus H, Bloch-
Zupan A, Carlos R, Carr IM, Downey LM, Blain KM, Mansfield DC, Shahrabi M,
Heidari M, Aref P, Abbasi M, Michaelides M, Moore AT, Kirkham J, Inglehearn CF.
Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive
cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet. 2009
Feb;84(2):266-73. doi: 10.1016/j.ajhg.2009.01.009.
34: Downey LM, Keen TJ, Jalili IK, McHale J, Aldred MJ, Robertson SP, Mighell A,
Fayle S, Wissinger B, Inglehearn CF. Identification of a locus on chromosome
2q11 at which recessive amelogenesis imperfecta and cone-rod dystrophy
cosegregate. Eur J Hum Genet. 2002 Dec;10(12):865-9. doi:
10.1038/sj.ejhg.5200884.
Who we are
Group Leads
Team members
Dr Ummey Hany u.hany@leeds.ac.uk
Ms Alice Rigby a.l.rigby@leeds.ac.uk
Mr Cheuk Wang Au c.w.au@leeds.ac.uk
Links
- Genomics Facility
- Aire (Advanced Research Computing)
- Leeds Centre for Disease Models
- Leeds Biomineralisation Research Group
- Genomic England Clinical Amelogenesis Imperfecta panel app
- The UK Amelogenesis Imperfecta Clinical Excellence Network
- Comprehensive review of AI genetics; Amelogenesis imperfecta: Genes, proteins and pathways
- SMILE AIDER (Stakeholder Meaningful InvoLvement and Engagement Aiding Dental Research) oral health patients and public involvement forum
- D3 (Developmental Dental Defects) group
- Leeds Dental School
- Thackray Medical Museum